Phát hiện trẻ mang đột biến gen tan máu bẩm sinh từ triệu chứng da nhợt nhạt
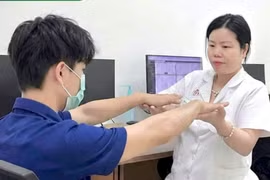
Bé trai B.N.M. (19 tháng tuổi, quê Phú Thọ) được gia đình đưa đến Bệnh viện Đa khoa tỉnh khám vì da nhợt nhạt kéo dài và chậm tăng cân so với trẻ cùng độ tuổi.
Theo gia đình, trẻ biểu hiện da nhợt nhạt khởi phát dần dần từ sau sinh, triệu chứng diễn tiến âm thầm, kéo dài. Trẻ sinh thường đủ tháng, quá trình mang thai và sinh nở không ghi nhận biến cố đặc biệt, chưa ghi nhận dị tật bẩm sinh, chưa từng truyền máu, phát triển vận động và tinh thần tương đối phù hợp với lứa tuổi, không có dấu hiệu chậm phát triển trí tuệ. Trẻ ăn uống được nhưng tăng cân chậm, chưa từng thiếu máu cấp hoặc nhập viện cấp cứu.
Sau đó, bệnh nhi được gia đình đưa đến Bệnh viện Medlatec để làm các xét nghiệm điện di huyết sắc tố và xét nghiệm gen Thalassemia.
Kết quả xét nghiệm, sắt huyết thanh nằm trong giới hạn bình thường. Điện di huyết sắc tố phát hiện HbH và Hb Bart’s, đồng thời HbA2 giảm, gợi ý đến bệnh Thalassemia thể α (Alpha). Xét nghiệm gen phát hiện đột biến gen dị hợp tử kép --SEA/-α3.7 trong bệnh α-Thalassemia.
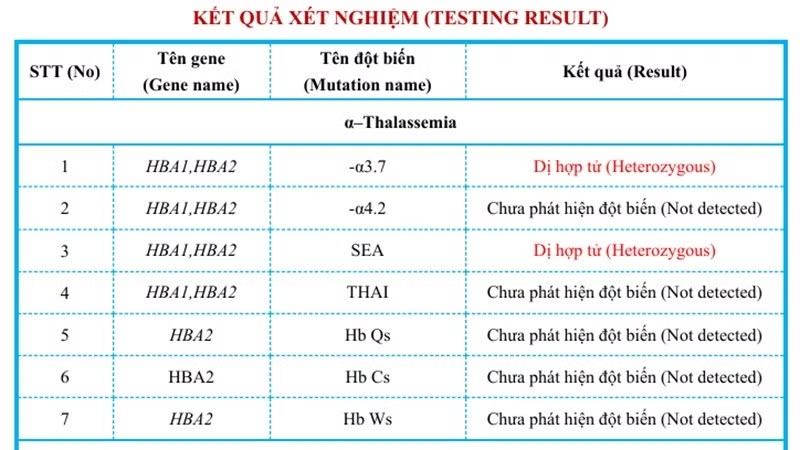
Kết quả xét nghiệm gen của bệnh nhân phát hiện đột biến gen dị hợp tử kép --SEA/-α3.7/Ảnh Medlatec
Bệnh nhi được chẩn đoán mắc α-Thalassemia thể HbH. Hiện tại, tình trạng sức khỏe trẻ tương đối ổn định, thiếu máu ở mức độ vừa và chưa có chỉ định truyền máu thường quy. Trẻ sinh hoạt và phát triển vận động phù hợp với lứa tuổi, tuy nhiên có biểu hiện da nhợt và chậm tăng cân do tình trạng thiếu máu mạn tính.
Bác sĩ tư vấn có thể can thiệp để trẻ cải thiện thể trạng, chủ yếu bằng bổ sung acid folic và xây dựng chế độ dinh dưỡng phù hợp. Nếu theo dõi tốt và kiểm soát các yếu tố kèm theo, nhiều trẻ thể bệnh HbH vẫn phát triển thể chất chấp nhận được và sinh hoạt gần như bình thường.
Tuy nhiên, gia đình cần theo dõi sát tình trạng sức khỏe của trẻ, đặc biệt trong các đợt bệnh cấp và đưa trẻ đi khám sớm để được đánh giá, hội chẩn thêm các chuyên khoa (Nhi khoa, Huyết học) khi cần thiết. Việc theo dõi định kỳ giúp phát hiện sớm các biến chứng và tối ưu hóa điều trị hỗ trợ.
Kiểm soát Thalassemia - bí kíp để sống khỏe, ngăn ngừa bệnh tật cho thế hệ sau
Theo ThS.BSNT Trần Hiền - Phòng Y sinh học di truyền, Trung tâm Xét nghiệm Medlatec, Thalassemia là bệnh di truyền lặn trên nhiễm sắc thể thường, đặc trưng bởi sự suy giảm hoặc thiếu hụt quá trình tổng hợp chuỗi globin trong phân tử hemoglobin. Tùy loại chuỗi globin bị ảnh hưởng, bệnh được chia thành α-thalassemia (thiếu chuỗi α) và β-thalassemia (thiếu chuỗi β).
Tại Việt Nam, theo số liệu của Viện Huyết học – Truyền máu Trung ương, người mang gen Thalassemia có mặt ở tất cả các dân tộc với tỷ lệ chung khoảng 13,8%. Đáng chú ý, tỷ lệ mang gen α-Thalassemia đặc biệt cao ở một số dân tộc ít người như dân tộc Tày (20,79%), Thái (22,2%) và Mường (22,39%).
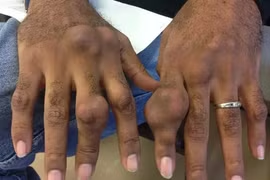
Khi mắc Thalassemia, hậu quả chính là thiếu máu mạn tính, khiến người bệnh thường xuyên mệt mỏi, da xanh, giảm khả năng học tập và lao động.
Ở các thể nặng, bệnh có thể gây gan lách to, biến dạng xương, chậm phát triển ở trẻ em, phải truyền máu định kỳ và dễ dẫn đến các biến chứng nghiêm trọng như suy tim, rối loạn nội tiết, giảm chất lượng cuộc sống. Đặc biệt, Thalassemia còn đe dọa sức khỏe lâu dài và khả năng sinh sản của thế hệ sau do nguy cơ di truyền bệnh cho con.
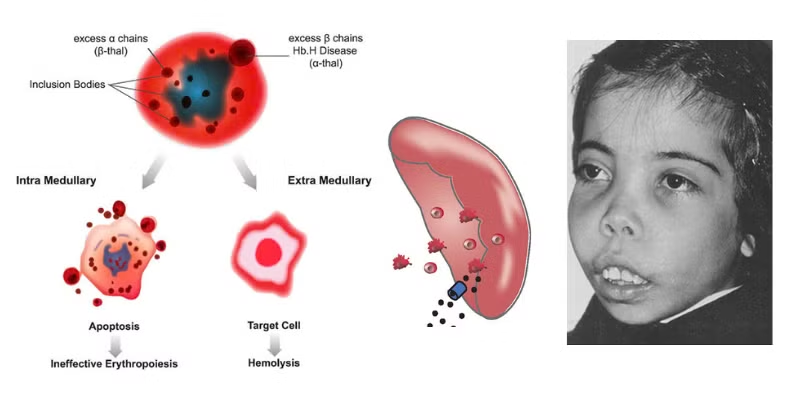
Thalassemia gây ra biến đổi trên cả tế bào máu và gương mặt người bệnh. Ảnh minh họa/Medlatec
Thalassemia không thể điều trị khỏi hoàn toàn bằng các biện pháp thông thường, tuy nhiên, bệnh có thể được kiểm soát hiệu quả nếu được chẩn đoán sớm và theo dõi đúng hướng.

![[INFOGRAPHIC] Trẻ em sử dụng thiết bị điện tử nguy hại gì?](https://cdn.kienthuc.net.vn/images/45c8388c62be8926eb035faa5fd7302d430b01db08d7e3da742c092d9d2e43281fddc5b3aabdce7e3086696b578dc7fa903c2dd3633e8d380245d5d0a4ffcb7ed4998af7f1cb8754e28e8b7cd13d3fed/thumb-tre-em-su-dung-thiet-bi-dien-tu.jpg.avif)